
Understanding Amyotrophic Lateral Sclerosis (ALS)
Amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig’s disease, is a rare neurological disease that affects motor neurons, those nerve cells in the brain and spinal cord that control voluntary muscle movement. Voluntary muscles are the ones we choose to move to produce movements such as chewing, walking and talking.
The disease is progressive, meaning that symptoms get worse over time. At present, no effective treatment has been found to reverse its progression.

ALS is a type of motor neuron disease. As motor neurons degenerate and die, they stop sending messages to the muscles, causing the muscles to weaken, begin to contract (contractions) and become lost (atrophy). Eventually, the brain loses its ability to initiate and control voluntary movements.
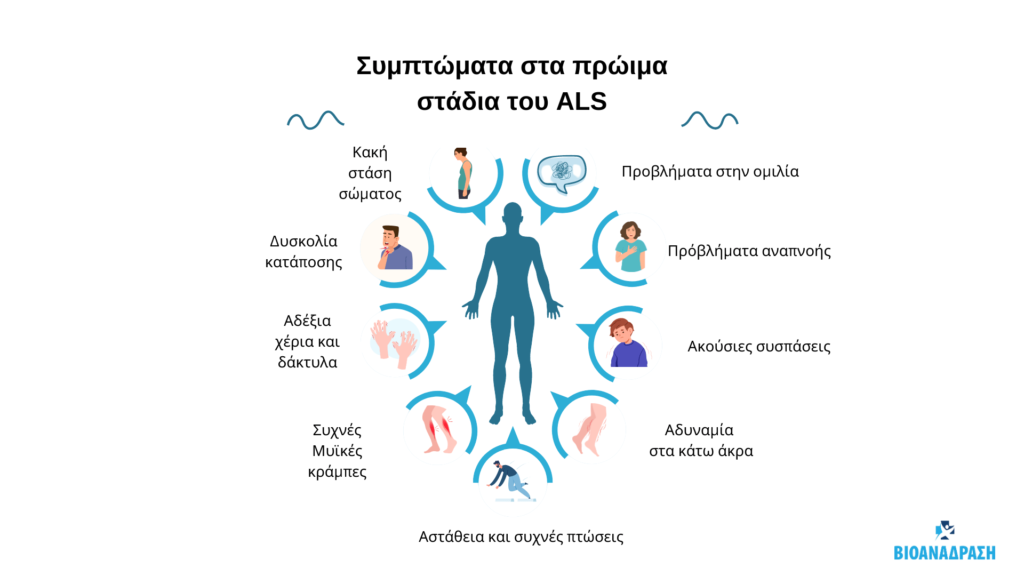
Early symptoms include:
- Muscle contractions in the arm, leg, shoulder or tongue
- Muscle cramps
- Tight and stiff muscles (spasticity)
- Muscle weakness affecting an arm, leg, neck or diaphragm
- Blurred and nasal speech
- Difficulty chewing or swallowing
As the disease progresses, muscle weakness and atrophy spread to other parts of the body.
Problems can develop with:
- Walking, carrying, getting out of bed on their own or using their hands and arms
- Chewing food and swallowing (dysphagia)
- Speaking or forming words (dysarthria)
- Breathing (shortness of breath)
- Weight maintenance and malnutrition
- Muscle cramps and neuropathy
- Anxiety and depression, because people with ALS usually remain able to reflect, remember, understand and be aware of their progressive loss of function
Diagnosis and treatment of amyotrophic lateral sclerosis
There is no test that can diagnose ALS for sure. The doctor in charge will perform a physical examination and review the complete medical history. A neurological examination will check reflexes, muscle strength and other responses and will be performed periodically to assess whether symptoms, such as muscle weakness, muscle loss and spasticity, are progressively worsening.
ALS treatment
So far, there is no cure for reversing the damage to motor neurons or treating ALS. However, treatments can make it easier to live with the disease.
Supportive healthcare is best provided by multidisciplinary teams of professionals such as doctors, pharmacists, physiotherapists, speech and language therapists, nutritionists, social workers, clinical psychologists and nurses.
These teams can design a personalised treatment plan and provide special equipment aimed at keeping people as mobile, comfortable and independent as possible.

Doctors can use the following drugs approved by the US Food and Drug Administration (FDA) to support a treatment plan for ALS:
- Riluzole (Rilutek) is an oral medication that is believed to reduce damage to motor neurons by reducing levels of glutamine, which carries messages between nerve cells and motor neurons.
- Edaravone (Radicava) is given intravenously and has been shown to slow the decline in physical activity of daily functions in people with ALS.
- Other medications may be prescribed to help manage symptoms such as muscle cramps, stiffness, excessive drooling and phlegm and unwanted episodes of crying and/or laughing or other emotional manifestations. Medications may also help when there is pain, depression, sleep disturbances and constipation.
Self-care or lifestyle changes
Nutritionists can provide guidance on how to plan and prepare small meals throughout the day that provide enough calories, fibre and fluids and how to avoid foods that are difficult to swallow.
If assisted feeding is impractical, doctors recommend the insertion of a feeding tube, which reduces the risk of choking and pneumonia that can result from inhaling fluids into the lungs.
Restoration
A treatment plan for ALS usually includes rehabilitation. Rehabilitation is different for everyone and a very important part of management.
Physiotherapy and Occupational Therapy
Physiotherapy plays an important role in maintaining the safety and independence of the person with ALS. Physical therapists are responsible in creating and recommending an individualized treatment program to maintain joint range of motion as well as strengthening muscles.
They may also recommend low-intensity exercises such as walking, swimming or using a stationary exercise bike. Caution! Extensive exercise can bring about the opposite effects, namely the destruction of already scarce muscle fibres.
Occupational therapists can recommend devices such as ramps, wheeled aids and wheelchairs to help save energy and maintain mobility.

Respiratory support
As the muscles responsible for breathing begin to weaken, there may be shortness of breath during physical activity and difficulty breathing at night. Non-invasive ventilation (NIV) refers to respiratory support usually provided through a mask over the nose and/or mouth. Initially, NIV may only be necessary at night, but may eventually be used full-time.
Because the muscles that control breathing become weak, there may also be difficulty in eliciting a loud cough. There are various techniques for increasing a loud cough, including mechanical cough assist devices.
As the disease progresses, mechanical ventilation (ventilators) may be needed to ventilate the lungs. Doctors may insert a breathing tube by mouth or surgically create a hole in the front of the throat and insert a tube leading into the windpipe (tracheostomy).
What is the difference between ALS and MS (Multiple Sclerosis)?
Multiple sclerosis (MS) and amyotrophic lateral sclerosis (ALS) are different diseases with similar features and symptoms.
Both:
- Affect the muscles and the ability to move the body
- Attacks the brain and spinal cord
- They have the word “hardening” in their nomenclature
- They cause scarring or hardening around nerve cells
However, they do have some key differences. MS is an autoimmune disease that causes the body to attack itself. ALS, also called Lou Gehrig’s disease, is a disorder of the nervous system that damages nerve cells in the brain and spinal cord. Both are treated differently.
Diseases and nerve cells
“Sclerosis-sclerosis” comes from the Greek word for “scar-scar”. Both ALS and MS cause scarring of the nerve fiber sheath, but the process of how this happens is different for each.
Nerve cells in the body are wrapped in thin caps called myelin sheaths. They protect these cells, similar to how insulation protects electrical wires. When you have MS, your body attacks the myelin sheaths in your brain and spinal cord.
When myelin sheaths are damaged, signals from the brain to other parts of the body are short-circuited.
ALS breaks down the actual nerve cells in the brain and spinal cord. These cells, called motor neurons, are responsible for voluntary muscles in the upper and lower limbs, face and diaphragm for breathing. Control of motor functions is lost and as the motor neurons break down, the myelin sheaths harden.

More differences
- MS is diagnosed earlier in life than ALS. It is usually discovered between the ages of 20 and 40. ALS is often diagnosed between 40 and 70.
- They affect the sexes differently. MS affects more women than men. ALS is more common in men.
- MS is more common in Caucasians. ALS affects all ethnic groups equally.
- ALS can be inherited, but MS cannot. Up to 10% of ALS cases are transmitted directly through genes. This is not the case with MS. But if your mom, dad or brother has multiple sclerosis, you’re at a higher risk of inheriting the disease.
- MS is more common than ALS.
- There is no cure for either disease, but treatments can help slow down both diseases. Lifestyle changes can help manage symptoms and improve quality of life.
References:
- The ALS association
- P. Masrori , P. Van Damme. (2020) Amyotrophic lateral sclerosis: a clinical review.Eur J Neurol. 7;27(10):1918-1929.Ryan G. Brotman; Maria C. Moreno-Escobar; Joe Joseph; Sunil Munakomi; Gauri Pawar. (2024) Amyotrophic lateral sclerosis.StatPearls Publishing.
By EVAGELIA MANTA, BSc, MT, IBITA Instructor, IKTA Instructor